
By Uwe Klein, PhD, Senior Vice President of Biological Sciences, Septerna, and Ryan Strachan, PhD, Principal Scientist, Discovery Biology, Septerna.
G-protein coupled receptors (GPCRs) are integral membrane proteins that mediate information transfer from the outside of a cell to the inside of a cell upon binding of a hormone, neurotransmitter, extracellular metabolite, odorant, drug, or other signal molecule (ligand). GPCRs are the largest family of receptors in the human genome and mediate a wide range of biological functions. Given their essential roles in human physiology and disease, GPCRs are effectively targeted by over a third of FDA-approved medications. However, the majority of GPCRs remain undrugged due to limitations imposed by conventional small molecule drug discovery approaches, challenges with biochemical studies and obtaining structures, and an incomplete understanding of their physiological roles. As presented recently at the 2nd Annual GPCRs-Targeted Drug Discovery Summit in Boston1, technologies like cryo-electron microscopy (cryo-EM), nanobodies, ultra-high throughput screens for small molecules and proteins, as well as conceptual frameworks such as allosteric modulation, have emerged to drive the development of next-generation GPCR targets and drugs. Here we focus on the common thread of allostery and how it can impact the future of GPCR drug discovery as it integrates with these emerging technologies.
Allosteric modulation is the process by which a molecule (or ligand) binds to one site on a receptor protein and changes the activity of a different site on the same protein2. Endogenous ligands – the naturally occurring binding partners of the receptor – engage well-conserved binding pockets commonly referred to as ‘orthosteric sites’ to modulate receptor function. Molecules that alter a protein’s function by binding outside these orthosteric sites are called allosteric modulators and work either in conjunction with or against the action of the co-bound endogenous agonist (Figure 1).
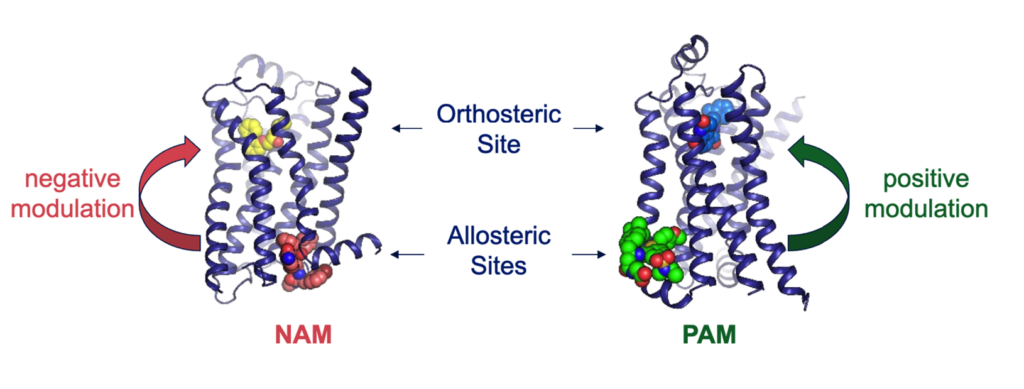
Figure 1. Orthosteric and allosteric sites on the β2-adrenergic receptor3
The concept of allosteric modulation is not new to the GPCR field4, though it has garnered major attention in the last 20 years as an emerging therapeutic modality having distinct advantages over conventional orthosteric agonists or antagonists that activate or inhibit a receptor’s function, respectively. Consequently, the field has seen a dramatic increase in the number of scientific publications covering allosteric modulation of GPCRs (Figure 2).
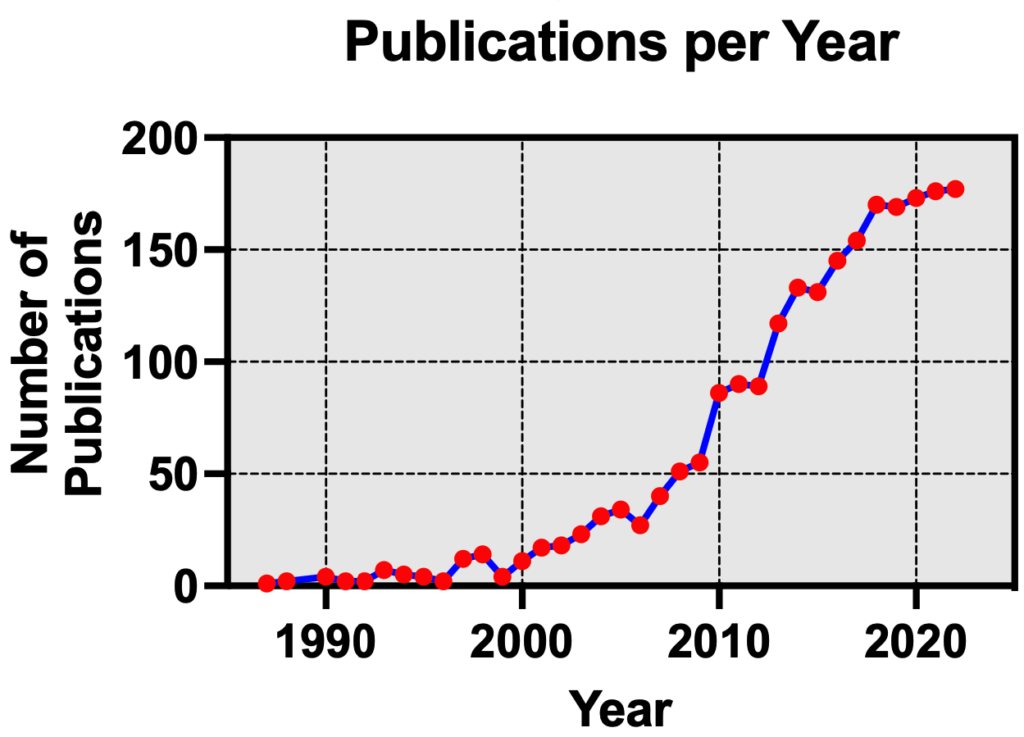
Figure 2. Number of publications each year covering allosteric modulation of GPCRs (PubMed search5)
Widely considered to be prototypic “allosteric machines”6, GPCRs adopt ligand-stabilized conformations within the plasma membranes of cells to relay extracellular stimuli to intracellular signaling cascades. According to this conventional mechanism, orthosteric agonist ligands stabilize active receptor conformations to elicit a cellular response. By contrast, orthosteric antagonists do not favor active conformations or even stabilize inactive conformations to block cellular responses. The recent explosion of GPCR structures solved by crystallography or cryo-EM shows that many additional allosteric sites – and thus many additional opportunities – exist for modulating GPCR function outside of more traditional orthosteric approaches7.
Allosteric modulation of GPCRs offers five key advantages over orthosteric activation or inhibition. First, the activity of allosteric modulators is saturable, meaning that they have a greater potential for fine-tuning physiological responses in either a positive or negative direction and have limits to safeguard against overdose. Second, for modulators that don’t elicit their own direct response, activity is only realized in the presence of an endogenous agonist, thereby preserving spatiotemporal aspects of physiologic signaling. Third, allosteric modulators do not affect all endogenous ligands in the same manner, thereby allowing for selective modulation of one ligand over another. Fourth, allosteric modulation can be biased, also termed “permissive”, which means that some allosteric modulators have disproportionate effects on the different downstream signaling pathways, thereby altering the natural output of a receptor. Finally, since allosteric binding sites are generally less evolutionarily conserved than orthosteric sites, selective effects on related receptor family members can be achieved with allosteric modulators.
The vast therapeutic potential of this novel concept for receptor modulation is already being realized within company pipelines as four GPCR allosteric modulators have gained FDA-approval, 25 are in clinical trials targeting 12 different GPCRs, and 340 are in preclinical development8.
In addition to small molecules, it is becoming evident that binding site-agnostic drug discovery approaches can identify ligands with novel mechanisms of action, such as large antibodies and nanobodies. Given that many of these molecules likely target sites other than the orthosteric binding pocket, it is incumbent on the field to understand the theory, advantages, and risks of allosteric modulation for successful outcomes. To this end, crafting allosteric modulator-specific drug discovery assays and critical paths for discovery are paramount to success. Hit identification needs to be tailored to the type of modulation desired for the therapeutic effect and any hits obtained need to be characterized for the specific mechanism of allosteric effect, strength of modulation, and binding affinity. Moreover, any potentially selective effects on a subset of ligands or signaling pathways need to be understood and de-risked preclinically. Taken together, a comprehensive understanding of core principles is required to realize the full therapeutic potential of GPCR allosteric modulation.
Septerna has embraced the concept of allosteric modulation in its discovery programs, recognizing the tremendous potential that exists in this new approach to GPCR therapeutic modulation. One example from our evolving pipeline is our Thyroid-Stimulating Hormone Receptor Negative Allosteric Modulator (TSHR NAM) program, targeting the treatment of patients with Graves’ disease and Thyroid Eye Disease. Both are disorders caused by an autoimmune response targeting the TSHR which gives rise to autoantibodies that cause aberrant receptor activation and thyroid hormone biosynthesis, leading to a hyperthyroid state that, in the case of Thyroid Eye Disease, is accompanied by ocular manifestations. Current treatment options are inefficient, poorly tolerated, or include radical surgery or radioablation. Considering the complex challenge of directly blocking TSHR activation by multiple autoantibodies with different binding sites, we have made great progress towards developing small molecule NAMs that leverage an allosteric site for broad inhibition of pathological TSHR signaling. Such a novel approach will be transformative for Graves’ patients.
Looking to the future, continued advances in high resolution structural techniques, computational chemistry, protein biochemistry, assay and screening technologies, and conceptual frameworks of allostery will further this new era of GPCR drug discovery that we are now in. By integrating fundamental concepts of GPCR allostery with innovative approaches to drug discovery, we will uncover the next generation of medicines for human diseases.
1 2nd Annual GPCRs-Targeted Drug Discovery Summit, May 23-25, 2023, Boston, MA, Conference Website.
2 Changeux JP and Christopoulos A (2016) Cell, 166(5):1084-1102.
3 Liu X et al. (2017) Nature, 548(7668):480-484; Liu X et al. (2019) Science, 364(6447):1283-1287.
4 De Lean A et al. (1980) J Biol Chem, 255(15):7108-17; Ehlert FJ (1988) J Pharmacol Exp Therap, 247(2):596-602; Fenalti G et al. (2014) Nature, 506(7487):191-6.
5 Results from PubMed Search on April 5, 2023, PubMed Search Link.
6 Kenakin TP (2012) Br J Pharmacol, 165(6):1659-1669.
7 Thal DM et al. (2018) Nature, 559(7712):45-53.
8 Persechino M et al. (2022) Pharmacology and Therapeutics, 237:108242.